- Get Started
- Centers
- Administrative Coordinating Center (ACC)
- Informatics Research Center (IRC)
- NIH
- Omics Centers
- Baylor Human Genome Sequencing Center
- Baylor-UTHealth Metabolomics Center
- Broad Institute Genomics Platform
- Broad Institute Metabolomics Platform
- Broad Institute and Beth Israel Proteomics Platform
- Illumina
- Keck MGC
- New York Genome Center Genomics
- Northwest Genomics Center
- New York Genome Center RNA-seq
- Psomagen
- Projects/Studies
- Working Groups
- Data
- Publications
- EEP
- ELSI
- Workshops
About TOPMed
Updated 10/24/2023
Contents
Overview
The Trans-Omics for Precision Medicine (TOPMed) program, sponsored by the National Institutes of Health (NIH) National Heart, Lung and Blood Institute (NHLBI), is part of a broader Precision Medicine Initiative, which aims to provide disease treatments tailored to an individual’s unique genes and environment. TOPMed contributes to this Initiative through the integration of whole-genome sequencing (WGS) and other omics (e.g., metabolic profiles, epigenomics, protein and RNA expression patterns) data with molecular, behavioral, imaging, environmental, and clinical data.
Study Characteristics
A primary goal of the TOPMed program is to improve scientific understanding of the fundamental biological processes that underlie heart, lung, blood, and sleep (HLBS) disorders. TOPMed is providing deep WGS and other omics data to pre-existing ‘parent’ studies having large samples of human subjects with rich phenotypic characterization and environmental exposure data.
Study Designs
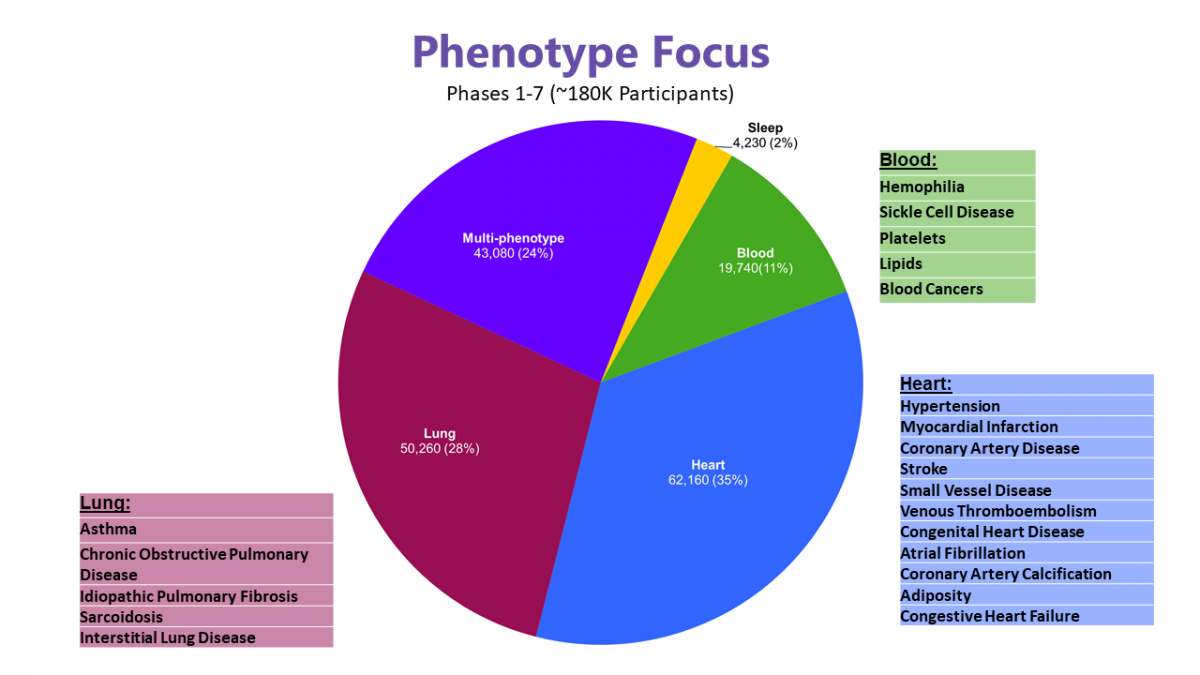
As of September 2021, TOPMed consists of ~180k participants from >85 different studies with varying designs. Prospective cohorts provide large numbers of disease risk factors, subclinical disease measures, and incident disease cases; case-control studies provide large numbers of prevalent disease cases; extended family structures and population isolates provide improved power to detect rare variant effects. The phenotype pie chart below shows the numbers and percentages of participants in studies with a focus on HLBS, as well as the percentage belonging to cohort studies that have collected many different phenotypes. It also shows areas of focus within each of the major HLBS categories.
Participant Diversity
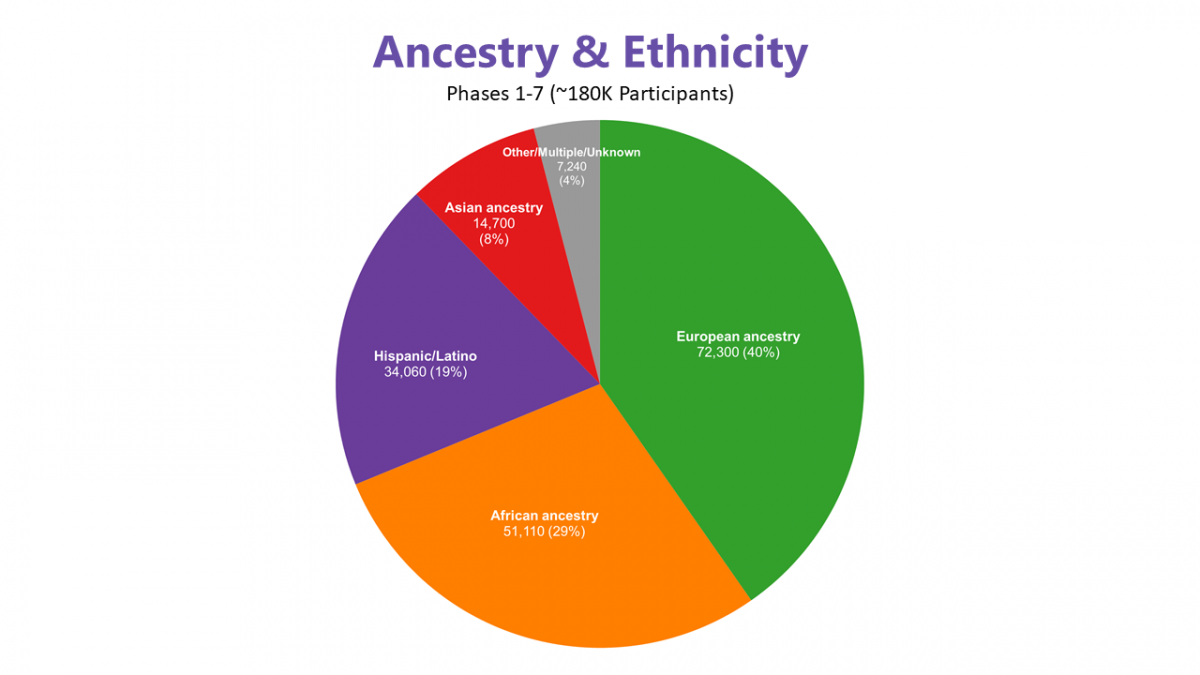
Achieving ancestral and ethnic diversity is a priority in selecting contributing studies. Currently, 60% of the 180k sequenced participants are of predominantly non-European ancestry. Discovery of genotype-phenotype associations frequently includes pooled analysis across ancestry groups and studies, using statistical models that account for population structure and relatedness.
The pie chart below summarizes TOPMed participant diversity using a combination of self-identified or ascriptive race/ethnicity categories, study inclusion criteria, or other demographic information provided by study investigators. Please note that while groupings may correlate to some extent with genetic ancestry, TOPMed recommends distinguishing between genetically and non-genetically inferred descriptions in analyses and publications, as described in these Guidelines on the use and reporting of race, ethnicity, and ancestry in TOPMed.
Whole Genome Sequencing
WGS was performed by several sequencing centers to a median depth of 30X using DNA from blood, PCR-free library construction and Illumina HiSeq X technology. A Support Vector Machine quality filter was trained with known variants and Mendelian-inconsistent variants. The Informatics Research Center conducts joint genotype calling across all samples available to produce genotype data “freezes.”
In TOPMed data freeze 8, with variant discovery on ~186k samples, 811 million single nucleotide variants and 66 million short insertion/deletion variants were identified and passed variant QC.
In TOPMed data freeze 9, variant discovery was initially made on ~206k samples including CCDG, but subset to 158,470 TOPMed samples plus 2,504 1000 Genomes samples. 781 million single nucleotide variants and 62 million short insertion/deletion variants were identified and passed variant QC. These variant counts are slightly smaller than the corresponding numbers in data freeze 8 due to omitting sites which show no variation in TOPMed samples. More information about WGS methods can be found under Sequencing and Data Processing Methods.
Omics
TOPMed Omics data processing is being performed by several sequencing centers. The program requires that omics data be submitted to dbGaP, along with thorough documentation of biosampling and laboratory methods, as well as sample provenance. Visit the Standards webpage to find available documented omics pipelines specific to omics type and phase. Below is a summary of the approved data sources for each study/cohort name categorized by data type.
TOPMed WGS and Omics Summary of Approved Projects |
|||||||||
| Short Name | Study/Cohort name | PI | Populations | dbGaP ID | WGS | RNA-seq | Methylation | Metabolomics | Proteomics |
|---|---|---|---|---|---|---|---|---|---|
| ATGC | Asthma Translational Genomics Collaborative | Burchard Esteban; Williams, L. Keoki; | ATGC dbGaP IDs | 16,494 | 9,290 | ||||
| MESA | Multi-Ethnic Study of Atherosclerosis | Rotter, Jerome; Rich, Stephen | Multi-ethnic populations | phs001416 | 7,107 | 8,903 | 13,286 | 12,800 | 14,200 |
| HCHS_SOL | Hispanic Community Health Study - Study of Latinos | Kaplan, Robert; North, Kari | phs001395 | 7,834 | 7,733 | 13,000 | 12,226 | ||
| ARIC+VTE | Venous Thromboembolism project | Boerwinkle, Eric | 20% African American | phs001211 phs001402 phs000993 | 10,531 | 6,111 | 16,524 | 16,524 | |
| CARDIA | Cell Disease Whole Genome Sequence Analysis in Early Cerebal Small Vessel Disease | Fomage, Myriam; Hou Lifang | phs001612 | 3,472 | 6,000 | 9,480 | 12,000 | 12,000 | |
| MLOF | My Life, Our Future: Genotyping for Progress in Hemophilia | Konkle, Barbara; Johnsen, Jill | phs001515 | 5,670 | 4,500 | ||||
| PVDOMICS | Pulmonary Vascular Disease Omics Analyses | Erzurum, Serpil; Barnard, John; Geraci, Mark; Beck, Gerald; Comhair, Suzy | phs002358 | 1,137 | 4,388 | 1,800 | |||
| SPIROMICS | SubPopulations and InteRmediate Outcome Measures In COPD Study | Meyers, Deborah A | phs001927 | 2,711 | 3,980 | 3,417 | |||
| FHS | Framingham Heart Study | Ramachandran, Vasan S.; Levy, Dan; Heard-Costa, Nancy | 3 generation EA pedigrees | phs000974 | 7,077 | 5,071 | 4,012 | 6,742 | 5,802 |
| Africa6K | Integrative Genomic Studies of Heart and Blood Related Traits in Africans | Tishkoff, Sarah; Williams, Scott | phs002194 | 6,392 | 2,934 | ||||
| HIPS | Hemophilia Inhibitor PUPs study | Brown, Deborah | phs002302 | 25 | 2,596 | ||||
| WHI | Women's Health Initiative | Kooperberg, Charles; Reiner, Alex | phs001237 | 11,310 | 2,365 | 4,400 | 4,400 | 1,000 | |
| LTRC | Lung TIssue Research Consortium | Silverman, Edwin | phs001662 | 1,541 | 1,548 | 3,041 | 1,548 | 1,548 | |
| IPF | Whole Genome Sequencing in Familial and Sporadic Idiopathic Pulmonary Fibrosis | Schwartz David; Fingerlin, Tasha | phs001607 | 2,883 | 835 | ||||
| PharmHU | The Pharmacogenomics of Hydroxyurea in Sickle Cell Disease | Boerwinkle, Eric; Sheehan, Vivien; Pace, Betty Sue | phs001466 | 862 | 826 | ||||
| COPDGene | Genetic Epidemiology of COPD | Silverman, Edwin | 30% African American | phs000951 phs000946 | 10,829 | 800 | 11,843 | 8,353 | |
| TOPCHeF | Trans-Omics for Precision Medicine for Congestive Heart Failure | Taylor, Matthew; Mestroni, Luisa; Graw, Sharon | phs002038 | 839 | 776 | 1,174 | |||
| nuMoM2b-HHS | nuMoM2b-Heart Health Study | Blue, Nathan; McNeil, Becky | 4,341 | 600 | |||||
| MDS | Genomics of Myelodysplastic Syndromes | Walter, Matthew; Goll, Johannes; Lindsley, R. Coleman; Saber, Wael; Padron, Eric; Miller, Christopher | phs002360 | 473 | 145 | ||||
| SCVI | Stanford Cardiovascular Institute iPSC Biobank Study | Wu, Joseph; Bustamante, Carlos | phs002338 | 1,163 | 82 | ||||
| AA_CAC | African American Coronary Artery Calcification project | Taylor, Kent D.; Rotter Jerome | African American Families | phs002194 | 1,159 | ||||
| AFGen | Identification of Common Genetic Variants for Atrial Fibrillation and PR Interval - Atrial Fibrilation Genetics Consortium | Ellinor, Patrick | AFGen dbGaP IDs | 12,742 | |||||
| Amish | Genetics of Cardiometabolic Health in the Amish | Mitchell, Braxton D. | Old Order Amish large extended pedigrees | phs000956 | 1,120 | ||||
| PGX_Asthma | Pharmacogenomics of Bronchodilator Response in Minority Children with Asthma | Burchard, Esteban; Hernandez, Ryan | 500AA, 500 Puerto Rican, and 500 Mexican of extremely non-responding asthma patients. | Please see this TOPMed Project's Parent Studies | 1,500 | ||||
| BAGS | Barbados Asthma Genetics Study | Barnes, Kathleen | African descent Barbados families with >40% of asthmatic members | phs001143 | 1,085 | ||||
| BCC-PREG | The Boston-Colombia Collaborative for Adverse Pregnancy Outcomes | Gray, Kathryn J.; Casa Romero, Juan P | Please see this TOPMed Project's Parent Studies. | 14,615 | |||||
| BioMe | Mount Sinai BioMe Biobank | Loos, Ruth J.F.; Kenny, Eimear | phs001644 | 11,626 | |||||
| Boston-Brazil_SCD | Boston-Brazil Collaborative Study of Sickle Cell Disease | Sankaran, Vijay G. | phs001599 | 415 | |||||
| CFS | Cleveland Family Study | Redline, Susan | African American | phs000954 | 1,300 | ||||
| CHS | Cardiovascular Health Study | Psaty, Bruce; Tracy, Russell | phs001368 | 4,780 | 8,041 | 8,619 | 8,619 | ||
| COPDMet | Plasma and BALF Metabolomics in COPDGene and SPIROMICS | Bowler, Russell | Please see this TOPMed Project's Parent Studies | 0 | |||||
| CRA_CAMP | The Genetic Epidemiology of Asthma in Costa Rica and the Childhood Asthma Management Program | Weiss, Scott T | Costa Rica is a special Hispanic population with asthma prevalence at 24% | phs001726, phs000988 | 6,647 | 3,000 | 3,000 | ||
| DS_CHD | Down Syndrome Associated Atrioventricular Septal Defects: New Omic Resources | Sherman, Stephanie L. | Please see this TOPMed Project's Parent Studies. | 469 | |||||
| ECLIPSE | Evaluation of COPD Longitudinally to Idenity Predictive Surrogate Endpoints | Silverman, Edwin | phs001472 | 2,355 | |||||
| GEM-OSA | Genetics, Epigenetics and Metabolomics of OSA subtypes | Pack, Allan; Carrier, Julie; Magalan, Ulysses; Mignot, Emmanuel; Ayas Najib | Please see this TOPMed Project's Parent Studies. | 3,000 | 3,000 | 3,000 | |||
| GeneSTAR | Genetic Studies of Atherosclerosis Risk | Mathias, Rasika | African American families, European families | phs001218 | 1,780 | ||||
| GenSalt | Genetic Epidemiology Network of Salt Sensitivity | He, Jiang | phs001217 | 1,858 | |||||
| GOLDN | Genetics of Lipid Lowering Drug and Diet Network | Amett, Donna K | European families | phs001359 | 965 | ||||
| HLKSCD | Genetic Variation of Heart, Lung, and Kidney Disease in Sickle Cell Disease: Pre- and Post- Curative Therapies | DeBaun, Michael; Eapen, Mary; Kang, Guolian; Edwards, Todd; Weiss, Mitch; Estepp, Jeremie; Gordeuk, Victor; Li, Bingshan; Saraf, Santosh | 1,780 | ||||||
| HyperGEN_GE NOA | Hypertension Genetic Epidemiology Network and Genetic Epidemiology Network of Arteriopathy | Amett, Donna K | African American families | phs001345 phs001293 | 3,153 | ||||
| JHS | Jackson Heart Study | Carson, April; Raffield, Laura | African American mixed family and population based | phs000964 | 3,418 | 1,659 | 5,266 | 5,266 | |
| OMG_SCD | Outcome Modifying Genes in Sickle Cell Disease | Ashley-Koch, Allison; Telen, Marilyn | phs001608 | 653 | |||||
| PCGC_CHD | Pediatric Cardiac Genomics Consortium's Congenital Heart Disease | Gelb, Bruce; Seidman, Christine | phs001735 | 3,888 | |||||
| PROMIS | Pakistan Risk of Myocardial Infarction Study | Saleheen, Danish | South Asian ancestry from Pakistan | phs001569 | 9,204 | ||||
| PUSH_SCD | Pulmonary Hypertension and the Hypoxic Response in Sickle Cell Disease | Nekhai, Sergei | phs001682 | 423 | |||||
| REDS-III_Brazil | Recipient Epidemiology and Donor Evaluation Study-III | Custer, Brian; Kelly, Shannon | Brazilian | phs001468 | 2,746 | ||||
| SAFS | San Antonio Family Studies | Blangero, John; Curran, Joanne | Mexican American in SAFHS extended pedigrees | phs001215 | 1,819 | ||||
| Samoan | Samoan Adiposity Study | McGarvey, Stephen | Samoan | phs000972 | 1,295 | ||||
| Sarcoidosis | Genetics of Sarcoidosis in African Americans | Montgomery, Courtney | African American families | phs001207 | 1,330 | ||||
| SARP | Severe Asthma Research Program | Meyers, Deborah A | phs001446 | 1,890 | |||||
| THRV | Taiwan Study of Hypertension using Rare Variants | Rotter, Jerome; Chen, Yii-Der Ida | Taiwan Chinese families | phs001387 | 2,170 | ||||
| UNID_CM | The Genetic Causes of Unexplained Cardiomyopathies | Seidman, Jonathan; Seidman, Christine | 779 | ||||||
| Walk-PHaSST | Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Therapy | Gladwin, Mark; Zhang, Yingze | phs001514 | 437 | |||||
| TOTAL | 205,092 | 69,483 | 94,260 | 97,895 | 48,435 | ||||
AFGen dbGaP IDs: phs001435, phs001543, phs001624, phs001732, phs001600, phs001189, phs001546, phs001606, phs001547, phs001725, phs001545, phs000993, phs001598, phs001062, phs001434, phs001544, phs001024, phs001601, phs001933, phs000997, phs001032, phs001040
ATGC dbGaP IDs: phs001728, phs001729, phs001730, phs001602, phs001603, phs001604, phs001605, phs000920, phs001542, phs001661, phs001727, phs000921, phs001467
Note: You may encounter phs links that redirect to a dbGaP error page in the table above. If so, this is because the TOPMed dbGaP study webpages do not go live until the study accession is released.
Note: TOPMed is generating a rich resource of multi-omics data that will include approximately 40K samples undergoing RNA-sequencing, 37K samples from metabolomics profiling, 57K samples from DNA methylation, and 4K samples from proteomics assaying. These projected totals include all stages of progress, from DNA/RNA that are currently being extracted, to those that are undergoing sequencing/profiling, or those that have completed the sequencing/profiling pipelines. Therefore, most omics data are in the process of being generated and will be released in the future.
Resources for the Scientific Community
TOPMed data are being made available to the scientific community as a series of “data freezes”: genotypes and phenotypes via dbGaP; read alignments via the Sequence Read Archive (SRA); and variant summary information via the Bravo variant server (see figure below) and dbSNP. Genotypes for a set of 55k samples have been released on dbGaP (freeze 5) and a freeze release of >140k samples is expected by mid 2020 (freeze 8). TOPMed WGS data are contained in study-specific accessions with names containing “NHLBI TOPMed”, while most phenotypic data are in parent study accessions. The TOPMed accessions can be identified by searching the dbGaP web site for “TOPMed”. More information about what data are available and how to access it can be found on the Data Access page.
TOPMed is currently adding other omic assays to samples that have been whole-genome sequenced; these include RNAseq, metabolomics, proteomics and epigenomics.
Overview of Bravo variant server resources
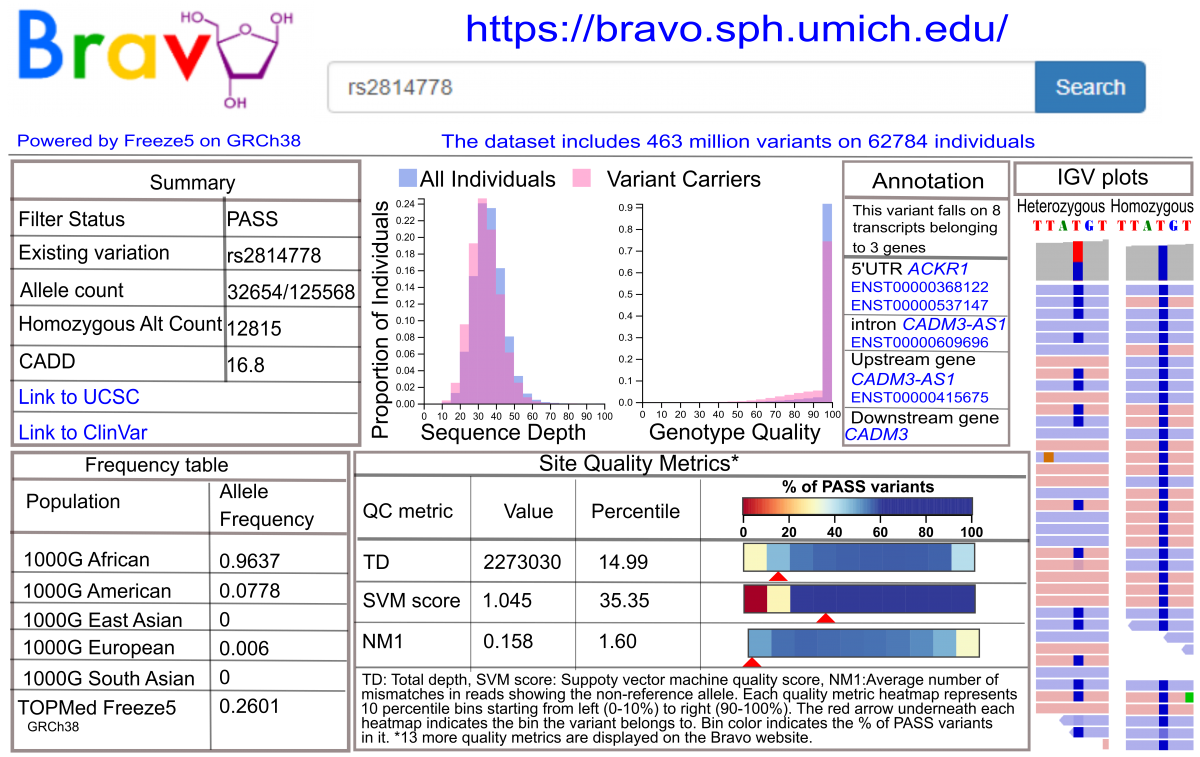
This content was adapted from a poster presented at the 2018 American Society of Human Genetics (ASHG) meeting, “Overview of the NHLBI Trans-Omics for Precision Medicine (TOPMed) program: Whole genome sequencing of >100,000 deeply phenotyped individuals” (Poster 3145/T).
SiteImprove

Building 31
31 Center Drive
Bethesda, MD 20892
1-877-NHLBI4U (1-877-645-2448)
Learn more about getting to NIH
Policies:
NHLBI Diversity, Equity and Inclusion Statement | Privacy Policy | Freedom of Information Act (FOIA) |
Accessibility | Copyright and Usage | HHS Vulnerability Disclosure | No FEAR Act | Grants and Funding
Customer Service/Center for Health Information | Email Alerts | Jobs and Careers | Site Index
About NHLBI | National Institutes of Health | Department of Health and Human Services | OIG | USA.gov